Rajca Research Group
Organic, Polymer and Biomaterials Chemistry
Rational Designs | Syntheses | Measurements
Many molecules and a few polymers with intrinsically chiral π-conjugated system are known. However, the traditional approach to π-conjugated polymers, directed at interesting material properties, is based on attaching chiral non-π-conjugated pendants to the approximately planar π-conjugated system of polymers and small donor/acceptor molecules. An alternate approach may rely on introducing π-conjugated chiral group, such as chiral binaphthyls or tetraphenylenes, as part of the polymer chain or network.
Organic conductors, superconductors, and magnets are based on electroactive π-conjugated molecules and polymers. For planar π-conjugated systems, the material properties are typically limited by weak intermolecular interactions in 3D. Transport properties (electrical conductivities, hole/electron mobilities) of organic molecules and polymers strongly depends on orientation/order of π-systems; in particular, π-stacking appears to play an essential role. Transition temperatures for organic magnets and superconductors were greatly improved with the advent of fullerene, replacing the pancake-like planar π-conjugated systems.
Typical π-stacks, involving planar π-conjugated molecules, are reminiscent of "stacks of pancakes".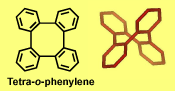 Tetra-o-phenylenes may be viewed as four such "pancakes" arranged in a double helical segment, rigidly fixed at 60 degree dihedral angles and pointing in four different directions. The tetraphenylene is derived from four phenylenes that are ortho-annelated to form an eight-membered ring in the center of the molecule. Although the tetraphenylene is an achiral, saddle-shape molecule with D2d point group of symmetry, substitution or annelation may break the symmetry, to provide chiral π-conjugated systems with extraordinary high barriers for racemiza-tion in the 60-70 kcal mol-1 range. These barriers are much higher than those for [n]helicenes.
Tetra-o-phenylenes may be viewed as four such "pancakes" arranged in a double helical segment, rigidly fixed at 60 degree dihedral angles and pointing in four different directions. The tetraphenylene is derived from four phenylenes that are ortho-annelated to form an eight-membered ring in the center of the molecule. Although the tetraphenylene is an achiral, saddle-shape molecule with D2d point group of symmetry, substitution or annelation may break the symmetry, to provide chiral π-conjugated systems with extraordinary high barriers for racemiza-tion in the 60-70 kcal mol-1 range. These barriers are much higher than those for [n]helicenes.
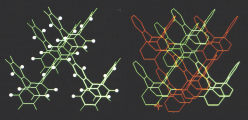 Succesive connections and annelations of tetraphenylenes would lead to a three-dimensional carbon network with the space group Pn3m, which was described by Riley in 1946. This carbon allotrope structure is of relatively low-strain, compared to fullerenes, and it remains experimentally elusive. Upon further analysis, we recognized that this achiral carbon allotrope could be dissected into π-conjugated double-helical polymers of tetraphenylenes, which may be viewed as two interwinding poly-m-phenylene helices or sequentially annelated tetraphenylenes.
Succesive connections and annelations of tetraphenylenes would lead to a three-dimensional carbon network with the space group Pn3m, which was described by Riley in 1946. This carbon allotrope structure is of relatively low-strain, compared to fullerenes, and it remains experimentally elusive. Upon further analysis, we recognized that this achiral carbon allotrope could be dissected into π-conjugated double-helical polymers of tetraphenylenes, which may be viewed as two interwinding poly-m-phenylene helices or sequentially annelated tetraphenylenes.
In 1997, we reported a convergent synthetic route to the racemic double helical octaphenylene, 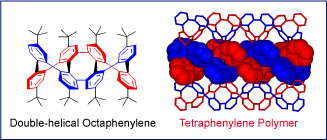 which is a fragment of a double helical polymer of tetraphenylenes (Angew. Chem. Int. Ed., 1997, 36, 488). The synthesis was based upon a stepwise connection and annelation of double helical fragments, the difunctionalized tetraphenylene. The octaphenylene was isolated in 4% yield and its structure was established by the detection of NMR ROESY cross-peaks between the protons that are brought close in space (distance of ~3.8 Å) by the double helical turn.
which is a fragment of a double helical polymer of tetraphenylenes (Angew. Chem. Int. Ed., 1997, 36, 488). The synthesis was based upon a stepwise connection and annelation of double helical fragments, the difunctionalized tetraphenylene. The octaphenylene was isolated in 4% yield and its structure was established by the detection of NMR ROESY cross-peaks between the protons that are brought close in space (distance of ~3.8 Å) by the double helical turn.
Our approach to the asymmetric synthesis of tetraphenylenes relies on the (-)-sparteine and copper dibromide mediated homocoupling of 2,2'-dilithiobiaryls. 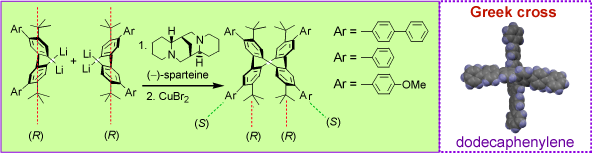 The formation of the tetraphenylene ring requires that the two chiral axes originating from 2,2'-dilithiobiaryls have identical configurations (e.g., (R)) but are opposite to the two new axes (e.g., (S)) that are associated with the newly formed CC bonds. Therefore, one of the prerequisites for high yields and high ee's is the facile configuration inversion of the biaryls on the experimental time scale, in the presence of (-)-sparteine. This synthetic methodology allowed us to prepare the dodecaphenylene, a molecule with two perpendicularly crossed hexaphenylenes which has the overall shape of a Greek cross (Tetrahedron, 2001, 57, 3725).
The formation of the tetraphenylene ring requires that the two chiral axes originating from 2,2'-dilithiobiaryls have identical configurations (e.g., (R)) but are opposite to the two new axes (e.g., (S)) that are associated with the newly formed CC bonds. Therefore, one of the prerequisites for high yields and high ee's is the facile configuration inversion of the biaryls on the experimental time scale, in the presence of (-)-sparteine. This synthetic methodology allowed us to prepare the dodecaphenylene, a molecule with two perpendicularly crossed hexaphenylenes which has the overall shape of a Greek cross (Tetrahedron, 2001, 57, 3725).
We reported the synthesis of tetranaphthylene, 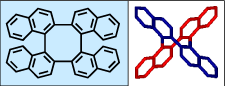 a chiral molecule with D2 point group of symmetry (J.Am. Chem. Soc., 2000, 122, 3351). Starting from (R)-(+)-2,2'-dibromo-1,1'-binapthyl, (R)-(+)-tetranaphthylene was obtained; its absolute configuration was recently established by professors Therese Friedman and Lawrence Nafie at Syracuse University, using vibrational circular dichroism spectroscopy (J. Phys. Chem. A, 2003, 107, 7692).
a chiral molecule with D2 point group of symmetry (J.Am. Chem. Soc., 2000, 122, 3351). Starting from (R)-(+)-2,2'-dibromo-1,1'-binapthyl, (R)-(+)-tetranaphthylene was obtained; its absolute configuration was recently established by professors Therese Friedman and Lawrence Nafie at Syracuse University, using vibrational circular dichroism spectroscopy (J. Phys. Chem. A, 2003, 107, 7692).
We also prepared the biphenylene dimers, a rather strained, planarized tetraphenylene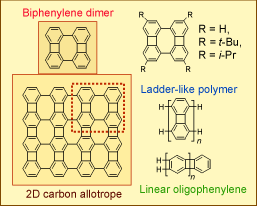 ( J. Am. Chem. Soc., 1996, 118, 7272). Such structure can be viewed as the molecular fragment of a 2D carbon allotrope, in which antiaromatic 4- and 8-membered rings are annelated with the aromatic 6-membered rings. The biphenylene dimer could also be viewed as a monomer for polymerization leading to double helical polymers of tetraphenylenes. In collaboration with the Rabinovitz group at Hebrew University, we reported the carbotetraanions of the biphenylene dimers, which were self-assembled with lithium cations forming helically stacked aggregates, including dimers, trimers, and tetramers.(J. Am. Chem. Soc., 2002, 124, 4685)
( J. Am. Chem. Soc., 1996, 118, 7272). Such structure can be viewed as the molecular fragment of a 2D carbon allotrope, in which antiaromatic 4- and 8-membered rings are annelated with the aromatic 6-membered rings. The biphenylene dimer could also be viewed as a monomer for polymerization leading to double helical polymers of tetraphenylenes. In collaboration with the Rabinovitz group at Hebrew University, we reported the carbotetraanions of the biphenylene dimers, which were self-assembled with lithium cations forming helically stacked aggregates, including dimers, trimers, and tetramers.(J. Am. Chem. Soc., 2002, 124, 4685)
Oligothiophenes have been intensively studied as materials for organic electronics. 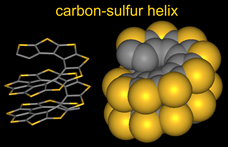 Much of the focus has been on α-oligothiophenes, in which thiophene rings are connected at the α-positions. Such oligothiophenes show excellent charge transport properties, thus have great potential as materials for organic field-effect transistors. It is known that succesive sequences of α-connection of thiophenes followed by β-annelation to form thiophene rings lead to a planar, quasi-linear extended conjugated π-system of carbon-sulfur oligomers.
Much of the focus has been on α-oligothiophenes, in which thiophene rings are connected at the α-positions. Such oligothiophenes show excellent charge transport properties, thus have great potential as materials for organic field-effect transistors. It is known that succesive sequences of α-connection of thiophenes followed by β-annelation to form thiophene rings lead to a planar, quasi-linear extended conjugated π-system of carbon-sulfur oligomers.
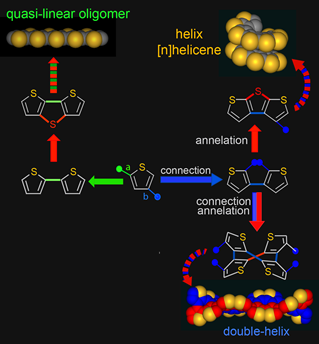 While exploring various possible helical structures, we recognized that, in contrast to the α-connection/β-annelation sequence, iterative sequences of β-connection of thiophenes and α-annelation to form thiophene ring would provide the esthetically pleasing carbon-sulfur helices. Similar to those for tetraphenylenes, the sequences of connections and connection/annelation of thiophenes would lead to double helical structures.
While exploring various possible helical structures, we recognized that, in contrast to the α-connection/β-annelation sequence, iterative sequences of β-connection of thiophenes and α-annelation to form thiophene ring would provide the esthetically pleasing carbon-sulfur helices. Similar to those for tetraphenylenes, the sequences of connections and connection/annelation of thiophenes would lead to double helical structures.
We developed the first synthesis of an annelated heptathiophene molecule, the carbon-sulfur [7]helicene, in which the thiophene rings are cross-conjugated and annelated into a (C2S)7 helix, a fragment of the unprecedented carbon-sulfur (C2S)n helix. ![carbon-sulfur [7]helicene](img/CS7Helix_gray.gif) (Angew. Chem. Int. Ed., 2000, 39, 4481). Our approach to the racemic synthesis of the [7]helicene was based upon two iterations, each consisting of a connection and annelation sequence. We reported asymmetric synthesis of the carbon-sulfur [7]helicene J. Am. Chem. Soc., 2004, 126, 15211). Learn more
(Angew. Chem. Int. Ed., 2000, 39, 4481). Our approach to the racemic synthesis of the [7]helicene was based upon two iterations, each consisting of a connection and annelation sequence. We reported asymmetric synthesis of the carbon-sulfur [7]helicene J. Am. Chem. Soc., 2004, 126, 15211). Learn more ![[7]helicene @ WordPress](img/wp1.png)
![carbon-sulfur [11]helicene](img/S11helix.png) We developed synthetic methodology for C-C bond forming cross-couplings between the β-positions of thiophenes in sterically hindered systems (Synlett, 2004, 177). Solving this difficult problem allowed us to implement asymmetric synthesis for relatively long fragments of the carbon-sulfur helix, substituted with alkyl chains (n-octyl groups) at the alpha-positions of the terminal thiophenes. For this series, our highest homologue to date is the undecathiophene, for which the structure was confirmed by X-ray crystallography (J. Am. Chem. Soc., 2005, 127, 13806).
We developed synthetic methodology for C-C bond forming cross-couplings between the β-positions of thiophenes in sterically hindered systems (Synlett, 2004, 177). Solving this difficult problem allowed us to implement asymmetric synthesis for relatively long fragments of the carbon-sulfur helix, substituted with alkyl chains (n-octyl groups) at the alpha-positions of the terminal thiophenes. For this series, our highest homologue to date is the undecathiophene, for which the structure was confirmed by X-ray crystallography (J. Am. Chem. Soc., 2005, 127, 13806).
Asymmetric synthesis of [11]helicene, employing a chiral additive such as (–)-sparteine in the final annelation step (forming one thiophene) and the final tri-annelation (forming three thiophenes), provides significant excess of left-handed and right-handed helices, respectively.
![Asymmetric Synthesis of carbon-sulfur [11]helicene](img/ASynthesis_S11helix2.bmp) The UV-vis absorbtion data indicated that the (C2S)n helix polymer had an optical band gap of Eg ≈ 3.5 eV and an onset of electron localization at n ≤ 7. The large Eg and early onset of localization were consistent with a cross-conjugated π-system for the carbon-carbon framework of (C2S)n helix polymer. These results were supported by electrochemical data; reversible cyclic voltammetric waves, corresponding to oxidation to radical cations, were found at similar potentials (1.2–1.3 V vs. SCE) for [7]helicenes and [11]helicene. In collaboration with the research group at the University of Málaga and Professor Rainer Glaser at the University of Missouri-Columbia, the vibrational spectroscopy studies on [7]helicene, [11]helicene, and annelated β-trithiophenes confirmed the cross-conjugated nature of the π-systems; density functional theory (DFT) computations suggested the presence of a nearly tetradegenerate HOMO in the α,α’-dimethyl-substituted [11]helicene, a model compound for dioctyl [11]helicene (J. Phys. Chem. C., 2007, 111, 4854; animation ). These results were in contrast to electron delocalization in other π-conjugated [n]helicenes, which possess much lower Eg = 2.1–2.5 eV. Learn more
The UV-vis absorbtion data indicated that the (C2S)n helix polymer had an optical band gap of Eg ≈ 3.5 eV and an onset of electron localization at n ≤ 7. The large Eg and early onset of localization were consistent with a cross-conjugated π-system for the carbon-carbon framework of (C2S)n helix polymer. These results were supported by electrochemical data; reversible cyclic voltammetric waves, corresponding to oxidation to radical cations, were found at similar potentials (1.2–1.3 V vs. SCE) for [7]helicenes and [11]helicene. In collaboration with the research group at the University of Málaga and Professor Rainer Glaser at the University of Missouri-Columbia, the vibrational spectroscopy studies on [7]helicene, [11]helicene, and annelated β-trithiophenes confirmed the cross-conjugated nature of the π-systems; density functional theory (DFT) computations suggested the presence of a nearly tetradegenerate HOMO in the α,α’-dimethyl-substituted [11]helicene, a model compound for dioctyl [11]helicene (J. Phys. Chem. C., 2007, 111, 4854; animation ). These results were in contrast to electron delocalization in other π-conjugated [n]helicenes, which possess much lower Eg = 2.1–2.5 eV. Learn more
Efficient syntheses of annelated β-trithiophenes (J. Org. Chem., 2006, 71, 3264), key building blocks for carbon-sulfur helicenes, will enable syntheses of the higher homologues of undecathiophene.
We reported the “helical fold-and-lock” concept in the synthesis of a bis[7]helicene with a rigid, helically locked structure (Angew. Chem. Int. Ed., 2009, 48, 5954). 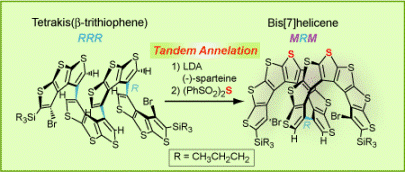 We demonstrated that the tetrakis(β-trithiophene) folded into a helical conformation (racemic RRR and SSS configurations) that facilitated the tandem annelation with high diastereoselectivity and modest enantioselectivity, to provide a bis[7]helicene with excess of MRM over PSP configuration. The enantioselectivity was mediated by (–)-sparteine. The rigid, helically locked structure of bis[7]helicene is imposed by the short intramolecular contacts between the curved π-systems of [7]helicene moieties, as revealed by synchrotron X-ray structure. The helically-locked bis[7]helicene possess chirooptical properties similar to those calculated for the corresponding long [n]helicene (n = 15). Learn more
We demonstrated that the tetrakis(β-trithiophene) folded into a helical conformation (racemic RRR and SSS configurations) that facilitated the tandem annelation with high diastereoselectivity and modest enantioselectivity, to provide a bis[7]helicene with excess of MRM over PSP configuration. The enantioselectivity was mediated by (–)-sparteine. The rigid, helically locked structure of bis[7]helicene is imposed by the short intramolecular contacts between the curved π-systems of [7]helicene moieties, as revealed by synchrotron X-ray structure. The helically-locked bis[7]helicene possess chirooptical properties similar to those calculated for the corresponding long [n]helicene (n = 15). Learn more
Although the solubility of the undecathiophene is excellent, we anticipate that for very long fragments of the carbon-sulfur helix, two terminal alkyl chains may not provide sufficient solubility. Therefore, we developed asymmetric synthesis for helicene building blocks incorporating solublizing groups. We prepared functionalized [7]helicene![functinalized [7]helicene](img/S6Bzhelix2.png) , in which a benzene ring functionalized with solubilizing n-heptyl chains is incorporated into the helix. This functionalization leads to another interesting phenomenon, i.e., the enantiomerically pure [7]helicene (e.g., helices with right-handed turn only) would not crystallize, forming rare chiral molecular glass. The racemate of the [7]helicene (a mixture of equal amounts with right- and left-handed helices) crystallizes, and its structure was confirmed by X-ray crystallography (Chem. Eur. J., 2004, 10, 6531). The design and synthesis of such chiral molecular glasses is important because the significant part of applications for organic materials, especially involving optics and optoelectronics, requires strictly isotropic glasses.
, in which a benzene ring functionalized with solubilizing n-heptyl chains is incorporated into the helix. This functionalization leads to another interesting phenomenon, i.e., the enantiomerically pure [7]helicene (e.g., helices with right-handed turn only) would not crystallize, forming rare chiral molecular glass. The racemate of the [7]helicene (a mixture of equal amounts with right- and left-handed helices) crystallizes, and its structure was confirmed by X-ray crystallography (Chem. Eur. J., 2004, 10, 6531). The design and synthesis of such chiral molecular glasses is important because the significant part of applications for organic materials, especially involving optics and optoelectronics, requires strictly isotropic glasses.
To enhance solubility, we prepared a new functionalized, enantiomerically pure [7]helicene . In this development, we implemented a modified synthetic route to the key building block, functionalized benzodithiophene, which could also serve as a versatile building block for organic-based electronic materials. Crystals of the functionalized benzodithiophene are chiral and adopt the shape of long, flexible, flat needles that can be readily bent.
. In this development, we implemented a modified synthetic route to the key building block, functionalized benzodithiophene, which could also serve as a versatile building block for organic-based electronic materials. Crystals of the functionalized benzodithiophene are chiral and adopt the shape of long, flexible, flat needles that can be readily bent. ![functinalized [7]helicene](img/S5Bz2Helix2.png) The new functionalized [7]helicene provided us a series of thiophene-based [7]helicenes with various degree electron delocalization to investigate a correlation between electron delocalization and chirooptical properties, which we found that chirooptical properties are not affected by electron delocalization in this series (J. Org. Chem., 2009, 74, 7504).
The new functionalized [7]helicene provided us a series of thiophene-based [7]helicenes with various degree electron delocalization to investigate a correlation between electron delocalization and chirooptical properties, which we found that chirooptical properties are not affected by electron delocalization in this series (J. Org. Chem., 2009, 74, 7504).
In the series of [7]helicenes available in our lab, we explored the effect of helix structure on intramolecular cyclization to provide a synthetic pathway to quasi-[8]circulenes![intramolecular cyclization of [7]helicene](img/quasicirculene.gif) . We found that the lack of atom linkage in the resultant quasi-[8]circulenes do not significantly affect properties and conformation, compared to those for the corresponding [8]circulenes. We note that the α-CH positions of quasi-[8]circulenes could enable functionalization, thus providing an avenue to cyclic structure of annelated aromatic rings with improved solubility and/or as building blocks for extended macromolecular structures and assemblies (J. Org. Chem., 2009, 74, 9105).
. We found that the lack of atom linkage in the resultant quasi-[8]circulenes do not significantly affect properties and conformation, compared to those for the corresponding [8]circulenes. We note that the α-CH positions of quasi-[8]circulenes could enable functionalization, thus providing an avenue to cyclic structure of annelated aromatic rings with improved solubility and/or as building blocks for extended macromolecular structures and assemblies (J. Org. Chem., 2009, 74, 9105).
In collaboration with Prof. Mieczyslaw Lapkowski at Silesian University (Poland), we investigated the first radical cation with chiral π-conjugated system,![Radical Cation of [7]helicene](img/radcat.png) radical cation of carbon−sulfur [7]helicene, that is configurationally stable in solution (J. Am. Chem. Soc., 2010, 132, 3246). The radical cation had a half life of 10 – 15 min in solution at room temperature. It was generated from the carbon−sulfur [7]helicene by spectroelectrochemistry and chemical oxidation, in which the progress of reactions were monitored by in situ UV-vis-NIR/CD/EPR spectroscoppy.
radical cation of carbon−sulfur [7]helicene, that is configurationally stable in solution (J. Am. Chem. Soc., 2010, 132, 3246). The radical cation had a half life of 10 – 15 min in solution at room temperature. It was generated from the carbon−sulfur [7]helicene by spectroelectrochemistry and chemical oxidation, in which the progress of reactions were monitored by in situ UV-vis-NIR/CD/EPR spectroscoppy.
Conjoined Double Helicene (J. Am. Chem. Soc., 2005, 127, 9312)
The conjoined helicene was discovered unexpectly during screening for the optimum routes to an annelated nitroxide diradical, derived from its coresponding diamine. Benzoyl peroxide (PhCO2)2 reacted with the planarized diamine, undergoing sequence of three oxidative CC- and NN-homocouplings (one CC and two NN), in which the two annelating cyclizations gave the chiral π-conjugated dihydrazine in high yield. X-ray structure of the dihydrazine crystal showed that the two [5]helicene-like fragments are annelated in their mid-sections, a conjoined double helicene structure. This synthetic pathway provides a novel, highly efficient approach to a chiral π-conjugated conjoined double helicene with remarkable configurational stability. Considering the molecular shape and the hydrazine moieties in the conjoined helicene, organic materials that are strongly chiral, electroactive, and isotropic may be envisioned. Resolution and syntheses of analogues of the hydrazine with extended conjoined helical structures are being pursued in our lab.
Benzoyl peroxide (PhCO2)2 reacted with the planarized diamine, undergoing sequence of three oxidative CC- and NN-homocouplings (one CC and two NN), in which the two annelating cyclizations gave the chiral π-conjugated dihydrazine in high yield. X-ray structure of the dihydrazine crystal showed that the two [5]helicene-like fragments are annelated in their mid-sections, a conjoined double helicene structure. This synthetic pathway provides a novel, highly efficient approach to a chiral π-conjugated conjoined double helicene with remarkable configurational stability. Considering the molecular shape and the hydrazine moieties in the conjoined helicene, organic materials that are strongly chiral, electroactive, and isotropic may be envisioned. Resolution and syntheses of analogues of the hydrazine with extended conjoined helical structures are being pursued in our lab.
Resources
Chirality @ UPenn
Chiral Information
Chirality: Description
Molecular Structures: Chirality
Chirality & Odour Perception
Carbon @ WebElement
Carbon Allotropes: Models
Allotropes of Carbon
Sulfur @ WebElement
Hamilton Hall 818 - 820,
Department of Chemistry,
University of Nebraska,
Lincoln, NE 68588-0304


