Rajca Research Group
Organic, Polymer and Biomaterials Chemistry
Rational Designs | Syntheses | Measurements








Our research is in the area of Organic Chemistry with emphasis on the design, synthesis and study of molecules with novel molecular structure and chemical properties. We are interested in fundamental aspects of electronic structure and its dependence on the size ranging from fraction of nanometer to several nanometers. The specific ongoing research projects are high-spin organic molecules and polymers as building blocks for organic magnets, organic radicals as contrast agents for biomedical imaging, and chiral π-conjugated molecules.
Organic radicals are typically highly reactive and unstable at ambient condition, as such their applications are very limited. The underlying theme of our research is the design and synthesis of organic radicals with improved stability and tailored properties for applications in novel magnetic materials and biomaterials.
Spotlights

![]()
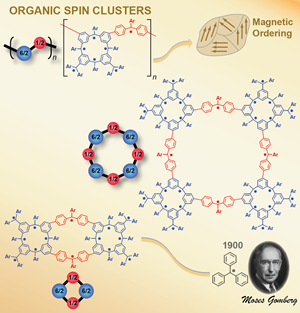 High-spin organic molecules and polymers are important to the understanding of organic magnetism and to the quest for organic magnets. We implemented the bottom-up approach based on the triphenylmethyl, the first organic free radical discovered by Moses Gomberg in 1900, in the design and synthesis of numerous high spin organic molecules and polymers
High-spin organic molecules and polymers are important to the understanding of organic magnetism and to the quest for organic magnets. We implemented the bottom-up approach based on the triphenylmethyl, the first organic free radical discovered by Moses Gomberg in 1900, in the design and synthesis of numerous high spin organic molecules and polymers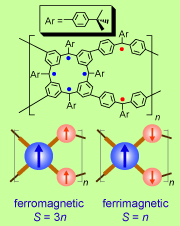 with dendritic and macrocyclic structures. Investigation of these high spin systems led to the concept of "organic spin cluster, a borne-in-failure concept that allowed for attaining very highspin polyradicals, and ultimately magnetic ordering in a polymer-based polyradical. We reported an organic π-conjugated polymer with very large magnetic moment (approximate average S = 5000) and magnetic order at low temperature (about 10 K). This is the first report of a π-conjugated organic polymer magnet. [Science, 2001, 294, 1503. Introduction to special issue: The Attraction of Magnetism] [2001 news] [Fermilab Colloquium]
with dendritic and macrocyclic structures. Investigation of these high spin systems led to the concept of "organic spin cluster, a borne-in-failure concept that allowed for attaining very highspin polyradicals, and ultimately magnetic ordering in a polymer-based polyradical. We reported an organic π-conjugated polymer with very large magnetic moment (approximate average S = 5000) and magnetic order at low temperature (about 10 K). This is the first report of a π-conjugated organic polymer magnet. [Science, 2001, 294, 1503. Introduction to special issue: The Attraction of Magnetism] [2001 news] [Fermilab Colloquium]
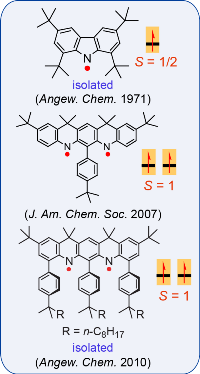 Nitrogen-centered (aminyl) radicals are typically short-lived, detected as intermediates in a variety of chemical and biological processes. Curious by the report on isolation of stable aminyl radical, tetra-tert-butylcarbazole aminyl, by Neugebauer and Fischer in 1971, we explored feasibilty of high spin aminyl radicals. In 2007, we reported the first high spin aminyl diradical that possesses triplet (S = 1) ground state (J. Am. Chem. Soc. 2007). The high spin aminyl is persistant at low temperature (half-life of 30 min at –70 °C). With improved molecular design, we prepared and isolated a triplet ground state aminyl diradical that
Nitrogen-centered (aminyl) radicals are typically short-lived, detected as intermediates in a variety of chemical and biological processes. Curious by the report on isolation of stable aminyl radical, tetra-tert-butylcarbazole aminyl, by Neugebauer and Fischer in 1971, we explored feasibilty of high spin aminyl radicals. In 2007, we reported the first high spin aminyl diradical that possesses triplet (S = 1) ground state (J. Am. Chem. Soc. 2007). The high spin aminyl is persistant at low temperature (half-life of 30 min at –70 °C). With improved molecular design, we prepared and isolated a triplet ground state aminyl diradical that 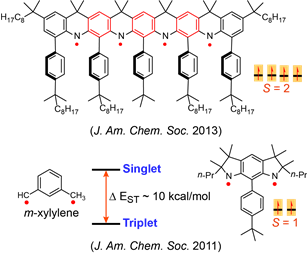 is persistent at room temperature and possesses a singlet-triplet energy gap that is significantly greater than the thermal energy at room temperature (Angew. Chem. Int. Ed. 2010). Recently, we reported high spin aminyl tetraradicals that have quintet (S = 2) ground states and persistent in solution at room temperature (J. Am. Chem. Soc. 2013).
is persistent at room temperature and possesses a singlet-triplet energy gap that is significantly greater than the thermal energy at room temperature (Angew. Chem. Int. Ed. 2010). Recently, we reported high spin aminyl tetraradicals that have quintet (S = 2) ground states and persistent in solution at room temperature (J. Am. Chem. Soc. 2013).
We also prepared another class of aminyl radicals that is a derivative of the well-known reactive intermediate m-xylylene diradical which possesses one of the largest singlet-triplet energy gaps (Δ EST ~ 9.6 kcal/mol) for an organic molecule. The planar derivative of aza-m-xylylene, aminyl diradical possesses a triplet ground state with Δ EST of about 10 kcal/mol. The aminyl diradical persists in solution at room temperature with half-life of 10 minutes (J. Am. Chem. Soc. 2011).
Ongoing research projects have the following specific objectives: (1) aminyl di- and triradicals with very strong pairwise ferromagnetic coupling between electron spins and persistence at room temperature with half-life that would permit isolation of the radicals, (2) aminyl polyradicals polymers with very large magnetic moment. Our long-term goal is a purely organic polymer with very large magnetic moment and magnetic order at room temperature.
Organic radicals can be sufficiently stabilized with adequate steric shielding of unpaired electron. Among very few examples of stable organic radicals is nitroxide, for which stability is also imparted by a three-electron bonding in the NO moiety. Because of their stability, nitroxides have a wide range of applications in chemistry, biochemistry and biomedicine. Note, however, that in vivo applications of nitroxides are rather limited, as they undergo fast reduction to the diamagnetic hydroxylamines.
In the search for stable polyradicals that are promising building blocks for preparation of organic polymer magnets ("plastic magnets"), we prepared and studied numerous nitroxides di- and polyradicals. 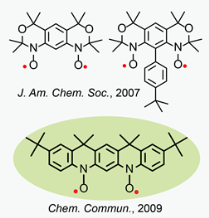 Among examples are high-spin nitroxide diradicals with conformationally-restricted structures, in which nitroxides are annelated to m-phenylene forming tricyclic benzobisoxazine-like structures (J. Am. Chem. Soc., 2007) and an annelated nitroxide diradical, analogue of the S = 1 aminyl diradical (Chem. Commun., 2009), which is stable in the solid state and in solution at ambient conditions. This is the first isolated nitroxide diradical with two diarylnitroxide moieties. Conformationally-constrained nitroxide polyradical scaffolds, such as calix[4]arene nitroxide radicals with fixed cone and 1,3 alternate conformations, were prepared and studied. The 1,3-alternate nitroxide diradical and tetraradical provide unique polyradical scaffolds for dissection of the through-bond and through-space intramolecular exchange couplings.
Among examples are high-spin nitroxide diradicals with conformationally-restricted structures, in which nitroxides are annelated to m-phenylene forming tricyclic benzobisoxazine-like structures (J. Am. Chem. Soc., 2007) and an annelated nitroxide diradical, analogue of the S = 1 aminyl diradical (Chem. Commun., 2009), which is stable in the solid state and in solution at ambient conditions. This is the first isolated nitroxide diradical with two diarylnitroxide moieties. Conformationally-constrained nitroxide polyradical scaffolds, such as calix[4]arene nitroxide radicals with fixed cone and 1,3 alternate conformations, were prepared and studied. The 1,3-alternate nitroxide diradical and tetraradical provide unique polyradical scaffolds for dissection of the through-bond and through-space intramolecular exchange couplings.
To explore feasibility of high-spin nitroxide di- and polyradicals (S > 1/2) 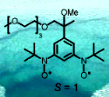 in biological and medical applications, we prepared a triplet ground state (S = 1) pegylated nitroxide diradical and, in collaboration with Professors Gareth and Sandra Eaton, we investigated the effect of water on its magnetic properties (Chem. Comm., 2005). This project has evolved to the synthesis of nitroxides for in vivo applications, in particular, development of biostable organic radicals as contrast agents (ORCAs) for magnetic resonance imaging and as spin labels for biophysical applications.
in biological and medical applications, we prepared a triplet ground state (S = 1) pegylated nitroxide diradical and, in collaboration with Professors Gareth and Sandra Eaton, we investigated the effect of water on its magnetic properties (Chem. Comm., 2005). This project has evolved to the synthesis of nitroxides for in vivo applications, in particular, development of biostable organic radicals as contrast agents (ORCAs) for magnetic resonance imaging and as spin labels for biophysical applications.
Nuclear magnetic resonance imaging (MRI) is one of the most powerful, non-invasive imaging tools for clinical diagnosis and screening. Paramagnetic materials are oftenly used in contrast-enhanced MRI scans to provide improved sensitivity and specificity. All MRI contrast agents currently in clinical use are metal-based, with gadolinium-based contrast agents (GBCAs) being the most widely used. Although most GBCAs are safe for the majority of patients, patients with renal insufficiency are at an increased risk for developing a serious systemic fibrosing disease (NSF) after receiving a GBCA [FDA Safety Warnings | FDA Questions and Answers on GBCAs]
Our goal is to develop of biostable organic radicals as contrast agent for MRI. Metal-free, organic-based agents would avoid the risk associated with toxic metal ions and provide an alternative to conventional paramagnetic agents.
The long-standing obstacle in the development of a practical organic radical contrast agent (ORCA) for MRI is the design and synthesis of paramagnetic organic compounds of moderate molecular size that possess sufficiently long in vivo lifetime, high water relaxivity, and high water solubility.
We successfully prepared an i.v injectable ORCA, 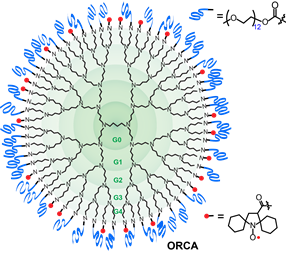 based on spirocyclohexyl nitroxide radicals and polyethylene glycol (PEG) chains conjugated to a generation 4 polypropylenimine (PPI) dendrimers scaffold, that has a long shelf‐life and provides selectively enhanced MRI in mice for over 1 h (J. Am. Chem. Soc., 2012). The large PPI-G4 allowed flexibility in adjusting the ratio of mPEGs and nitroxide to obtain the first ORCA that has a long shelf‐life and provides enhanced MRI in mice for over 1 h. The results demonstrate that ORCA can be designed to have high relaxivity to provide high resolution images of mouse organs. Molecular size of the ORCA based on PPI-G4 is at the upper size-limit for passage through the renal glomerular barrier, undergoes slow excretion through the kidneys. Optimization of ORCAs to acheive high water relaxivity, high water solubility and small, compact molecular size is the next crucial step in the development of ORCAs for clinical application.
based on spirocyclohexyl nitroxide radicals and polyethylene glycol (PEG) chains conjugated to a generation 4 polypropylenimine (PPI) dendrimers scaffold, that has a long shelf‐life and provides selectively enhanced MRI in mice for over 1 h (J. Am. Chem. Soc., 2012). The large PPI-G4 allowed flexibility in adjusting the ratio of mPEGs and nitroxide to obtain the first ORCA that has a long shelf‐life and provides enhanced MRI in mice for over 1 h. The results demonstrate that ORCA can be designed to have high relaxivity to provide high resolution images of mouse organs. Molecular size of the ORCA based on PPI-G4 is at the upper size-limit for passage through the renal glomerular barrier, undergoes slow excretion through the kidneys. Optimization of ORCAs to acheive high water relaxivity, high water solubility and small, compact molecular size is the next crucial step in the development of ORCAs for clinical application.
Nitroxides are widely used as spin labels for study of biomolecules. Site-directed spin labeling (SDSL) pulsed electron paramagnetic resonance (EPR) is among the best techniques for determination of conformational changes and for characterization of the flexible regions of macromolecules. In a typical approach, doubly-labeled macromolecules are prepared by site-directed spin labeling and a set of distances between spin labels is measured using pulsed electron paramagnetic resonance (EPR). The temperatures at which these measurements can be made are limited by the dynamic averaging effects associated with methyl group rotation in the conventional nitroxide spin label, and therefore have to be performed at about 50 – 65 K, requiring use of liquid helium and involve rigorous experimental setup.
Our goal is to designed and synthesize ultra-rigid, small-sized spin labels that would enable accurate, long-distance EPR distance measurements at temperatures significantly above 77 K (boiling point of liquid nitrogen) and ultimately at physiological temperatures.
We reported the synthesis and characterization of spirocyclohexyl nitroxide α-amino acid and its N-(9-fluorenylmethoxycarbonyloxy) (Fmoc) derivative 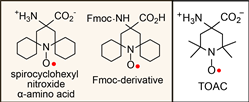 (Chem. Euro. J., 2010). The spirocyclohexyl spin label with rigid structure is an analogoue of 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4carboxylic acid (TOAC), the probe of choice for the study of proteins and peptides. We found that replacecing the two methyl groups in TOAC with rigid, spirocyclic cylohexane rings provided the spin label with long phase memory relaxation time that enables the distance measurements by EPR at liquid nitrogen temperature. Attempts to incorporate the Fmoc derivative of spirocyclohexyl spin label into peptides have been unsuccessful so far.
(Chem. Euro. J., 2010). The spirocyclohexyl spin label with rigid structure is an analogoue of 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4carboxylic acid (TOAC), the probe of choice for the study of proteins and peptides. We found that replacecing the two methyl groups in TOAC with rigid, spirocyclic cylohexane rings provided the spin label with long phase memory relaxation time that enables the distance measurements by EPR at liquid nitrogen temperature. Attempts to incorporate the Fmoc derivative of spirocyclohexyl spin label into peptides have been unsuccessful so far.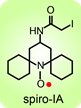 We prepared another derivative, spirocyclohexyl iodoacetamide (spiro-IA) spin label, which bind covalently with the thiol group of a cysteine residue. In collaboration with Professor Hassane Mchaourab at Vanderbilt University, spiro-IA labels have been sucessfuly incorpoared into T4 lysozyme (T4L). DEER measurements at room temperature have been sucessfuly performed on the spin labeled T4L in a disaccharide (trehalose) glassy matrix at University of Denver in the Eaton laboratory.
We prepared another derivative, spirocyclohexyl iodoacetamide (spiro-IA) spin label, which bind covalently with the thiol group of a cysteine residue. In collaboration with Professor Hassane Mchaourab at Vanderbilt University, spiro-IA labels have been sucessfuly incorpoared into T4 lysozyme (T4L). DEER measurements at room temperature have been sucessfuly performed on the spin labeled T4L in a disaccharide (trehalose) glassy matrix at University of Denver in the Eaton laboratory.
As a team member of the Protein Core at Membrane Protein Structural Dynamics Consortium (MPSDC), we lend expertise in organic radical chemistry and synthesis to the development of new protein expression protocols.
Chiral π-conjugated molecules are important building blocks in the the design of organic optoelectronic materials and devices. Highly annelated, chiral π-systems, such as [n]helicenes and tetraphenylenes, are known to possess strong chiral properties and high configurational stability, which are prerequisites for many chiral materials.
Helicenes, the structures with ortho-fused aromatic rings, are among the molecules with inherently strongest chiral properties, 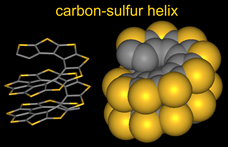 due to helical shape of their π-conjugated systems. Oligothiophenes have been intensively studied as materials for organic electronics. α-Oligothiophenes, in which thiophene rings are connected at the α-positions, have great potential as materials for organic field-effect transistors. Succesive sequences of α-connection of thiophenes followed by β-annelation to form thiophene rings lead to a planar, quasi-linear extended conjugated π-system of carbon-sulfur oligomers. In contrast to the α-connection/β-annelation sequence, iterative sequences of β-connection of thiophenes and α-annelation to form thiophene ring provide the esthetically pleasing carbon-sulfur helices.
due to helical shape of their π-conjugated systems. Oligothiophenes have been intensively studied as materials for organic electronics. α-Oligothiophenes, in which thiophene rings are connected at the α-positions, have great potential as materials for organic field-effect transistors. Succesive sequences of α-connection of thiophenes followed by β-annelation to form thiophene rings lead to a planar, quasi-linear extended conjugated π-system of carbon-sulfur oligomers. In contrast to the α-connection/β-annelation sequence, iterative sequences of β-connection of thiophenes and α-annelation to form thiophene ring provide the esthetically pleasing carbon-sulfur helices.
We developed the first synthesis of an annelated heptathiophene molecule, the carbon-sulfur [7]helicene, in which the thiophene rings are cross-conjugated and annelated into a (C2S)7 helix, a fragment of carbon-sulfur (C2S)n helix. ![carbon-sulfur [7]helicene](img/CS7Helix_gray.gif) (Angew. Chem. Int. Ed., 2000. The approach to the racemic synthesis of the [7]helicene was based upon two iterations, each consisting of a connection and annelation sequence. In 2004, we reported asymmetric synthesis of the carbon-sulfur [7]helicene J. Am. Chem. Soc., 2004). We further developed asymmetric synthesis for relatively long fragments of the carbon-sulfur helix, substituted with alkyl chains (n-octyl groups) at the alpha-positions of the terminal thiophenes. The highest homologue prepared to date is the undecathiophene, for which the structure was confirmed by X-ray crystallography (J. Am. Chem. Soc., 2005).
(Angew. Chem. Int. Ed., 2000. The approach to the racemic synthesis of the [7]helicene was based upon two iterations, each consisting of a connection and annelation sequence. In 2004, we reported asymmetric synthesis of the carbon-sulfur [7]helicene J. Am. Chem. Soc., 2004). We further developed asymmetric synthesis for relatively long fragments of the carbon-sulfur helix, substituted with alkyl chains (n-octyl groups) at the alpha-positions of the terminal thiophenes. The highest homologue prepared to date is the undecathiophene, for which the structure was confirmed by X-ray crystallography (J. Am. Chem. Soc., 2005).
We reported the “helical fold-and-lock” concept in the synthesis of a bis[7]helicene with a rigid, 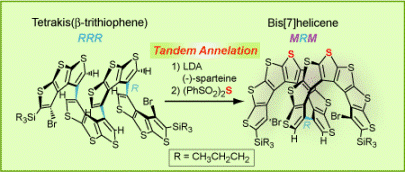 helically locked structure (Angew. Chem. Int. Ed., 2009). We demonstrated that the tetrakis(β-trithiophene) folded into a helical conformation (racemic RRR and SSS configurations) that facilitated the tandem annelation with high diastereoselectivity and modest enantioselectivity, to provide a bis[7]helicene with excess of MRM over PSP configuration. The enantioselectivity was mediated by (–)-sparteine. The rigid, helically locked structure of bis[7]helicene is imposed by the short intramolecular contacts between the curved π-systems of [7]helicene moieties, as revealed by synchrotron X-ray structure. The helically-locked bis[7]helicene possess chirooptical properties similar to those calculated for the corresponding long [n]helicene (n = 15).
helically locked structure (Angew. Chem. Int. Ed., 2009). We demonstrated that the tetrakis(β-trithiophene) folded into a helical conformation (racemic RRR and SSS configurations) that facilitated the tandem annelation with high diastereoselectivity and modest enantioselectivity, to provide a bis[7]helicene with excess of MRM over PSP configuration. The enantioselectivity was mediated by (–)-sparteine. The rigid, helically locked structure of bis[7]helicene is imposed by the short intramolecular contacts between the curved π-systems of [7]helicene moieties, as revealed by synchrotron X-ray structure. The helically-locked bis[7]helicene possess chirooptical properties similar to those calculated for the corresponding long [n]helicene (n = 15).
Typical π-stacks, involving planar π-conjugated molecules, are reminiscent of "stacks of pancakes". Tetra-o-phenylenes may be viewed as four such "pancakes" arranged in a double helical segment, 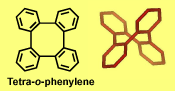 rigidly fixed at 60 degree dihedral angles and pointing in four different directions. The tetraphenylene is derived from four phenylenes that are ortho-annelated to form an eight-membered ring in the center of the molecule. Although the tetraphenylene is an achiral, saddle-shape molecule with D2d point group of symmetry, substitution or annelation may break the symmetry, to provide chiral π-conjugated systems with extraordinary high barriers for
rigidly fixed at 60 degree dihedral angles and pointing in four different directions. The tetraphenylene is derived from four phenylenes that are ortho-annelated to form an eight-membered ring in the center of the molecule. Although the tetraphenylene is an achiral, saddle-shape molecule with D2d point group of symmetry, substitution or annelation may break the symmetry, to provide chiral π-conjugated systems with extraordinary high barriers for 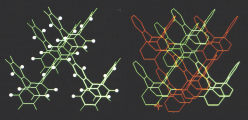 racemiza-tion in the 60-70 kcal mol-1 range. These barriers are much higher than those for [n]helicenes.
Succesive connections and annelations of tetraphenylenes would lead to a three-dimensional carbon network with the space group Pn3m, which was described by Riley in 1946. This carbon allotrope structure is of relatively low-strain, compared to fullerenes, and it remains experimentally elusive. This achiral carbon allotrope could be dissected into π-conjugated double-helical polymers of tetraphenylenes, which may be viewed as two interwinding poly-m-phenylene helices or sequentially annelated tetraphenylenes.
racemiza-tion in the 60-70 kcal mol-1 range. These barriers are much higher than those for [n]helicenes.
Succesive connections and annelations of tetraphenylenes would lead to a three-dimensional carbon network with the space group Pn3m, which was described by Riley in 1946. This carbon allotrope structure is of relatively low-strain, compared to fullerenes, and it remains experimentally elusive. This achiral carbon allotrope could be dissected into π-conjugated double-helical polymers of tetraphenylenes, which may be viewed as two interwinding poly-m-phenylene helices or sequentially annelated tetraphenylenes.
Hamilton Hall 818 - 820,
Department of Chemistry,
University of Nebraska,
Lincoln, NE 68588-0304


