Synthesis of stable organic diradicals
in an effort to maximize the energy between the singlet and triplet states.
By Joshua Schmidt
Contributors:
Dr. Andrzej Rajca
Teerapat Rojsajjakul
Dr. Jun Li
Natalie Ballard
Center for Materials Research and Analysis
Departments of Chemistry and Physics
University of Nebraska, Lincoln
Funded in Part By:
The National Science Foundation
Abstract:
This report outlines the methods used by me to synthesize one
stable acyclic diradical. In addition to this, it discusses the methods
for synthesizing a complementary cyclic diradical; both of which are important
to current research being performed on organic molecules with magnetic
properties. This includes the theory and reasoning behind the synthesis
of these two molecules and what information can be hoped to gain from them.
Along with that is also a discussion of possible uses and background behind
the research on some organic magnets.
Ó August 2000
Today's world is increasingly magnetic. From the simple compass to
superfast complex computers, and from credit cards to CAT scans and MRIs,
there is not a day we go about where we do not have some contact with magnets
and their applications. For this reason, many chemists are striving
to create new and innovative magnets using organic materials (carbon, oxygen,
nitrogen, and hydrogen). The possible applications for these types
of magnets are endless and as yet unknown; but possible uses include ambient
temperature superconductors, higher density data storage, MRI contrast
agents, and many others. This summer I was fortunate enough to be
involved in this research with the Rajca group at the University of Nebraska.
I was afforded this opportunity by a National Science Foundation grant
to the Research Experience for Undergraduates program supported by the
Center for Materials Research and Analysis at the University of Nebraska,
Lincoln. The following report is a short account of what I learned
and did while working in the Rajca lab.
Many years ago the first radical was observed and was noted to behave differently
than other molecules. This was because a radical is a molecule that
contains one or more unpaired electrons. All electrons have a spin
direction that is seen as a vector with a value of ½. When
in a magnetic field, we then assign them a relative value of ± ½
in relation to the direction of the field present. In normal molecules
all these electrons are paired with another electron of the opposite spin
in the same orbital giving the molecule a net spin (S) value of 0.
Therefore, if there is one extra electron the molecule will have a net
spin (S) value of ½. Because magnetism is caused by an excess
S in a molecule or atom, a molecule with S = ½ would be slightly
magnetic, but so slightly so that it is of no use anywhere. In an
attempt to raise the value of S, chemists have been trying to increase
the number of unpaired electrons on a molecule without having them pair
up and negate each other. This problem involves many factors including
how much the electrons interact and the resultant energy levels of the
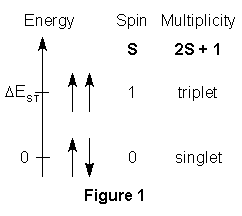
singlet and triplet states. The singlet state is when two radical
electrons are paired such that they have opposite spins and an S = 0, resulting
in a usually favored lower energy state in many molecules. The triplet
state is when two electrons are paired such that their spins are parallel
and S = 1, resulting in a usually unfavored higher energy state.
This is illustrated in Figure11. One effort of
current research is to increase the energy difference (DEST) between the
singlet and triplet states. Once we know we can control the energy
difference, we can design a molecule with the triplet state at a much lower
energy and thus give the molecule a high spin at ambient (room) temperature.
It has been theorized that by increasing the number of pathways between
the unpaired electrons the energy difference will increase by a factor
equal to the number of pathways2. To test this, two complementary
diradicals must be synthesized and tested.
Before beginning an explanation of the synthesis, a short explanation about
the multiple pathway theory is in order. In reference to Figure 2;
as is seen there is an exchange interaction value of J between the
two phenyl rings over the Exchange Coupling Pathway (ECP). Because
J = Jijeff
rirj
over
one ECP, where Jijeff is the effective exchange
integral between sites i and j, and ri and rj are the spin densities on
sites i and j, then over two pathways J = 2Jijeff
rirj.
This effectively doubles J in a two ECP system relative to a one
ECP system. Because the energy difference between the singlet and
triplet states is equal to 2J over one pathway, a doubling of pathways
would make the energy difference equal to 4J, or twice what it was
with only one pathway2.
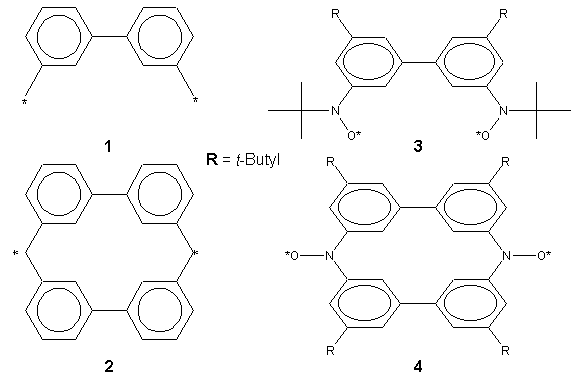
In order to test this, molecules such as 1 and 2 have been
synthesized, but both were so unstable they could only be worked on at
extremely low temperatures and could have no contact with air. It
was this reason that I was striving to create two molecules, 3 and
4, which were stable at room temperature and easy to handle in air.
Unfortunately, because of time constraints I was unable to synthesize 4
but was very successful synthesizing molecule 3.
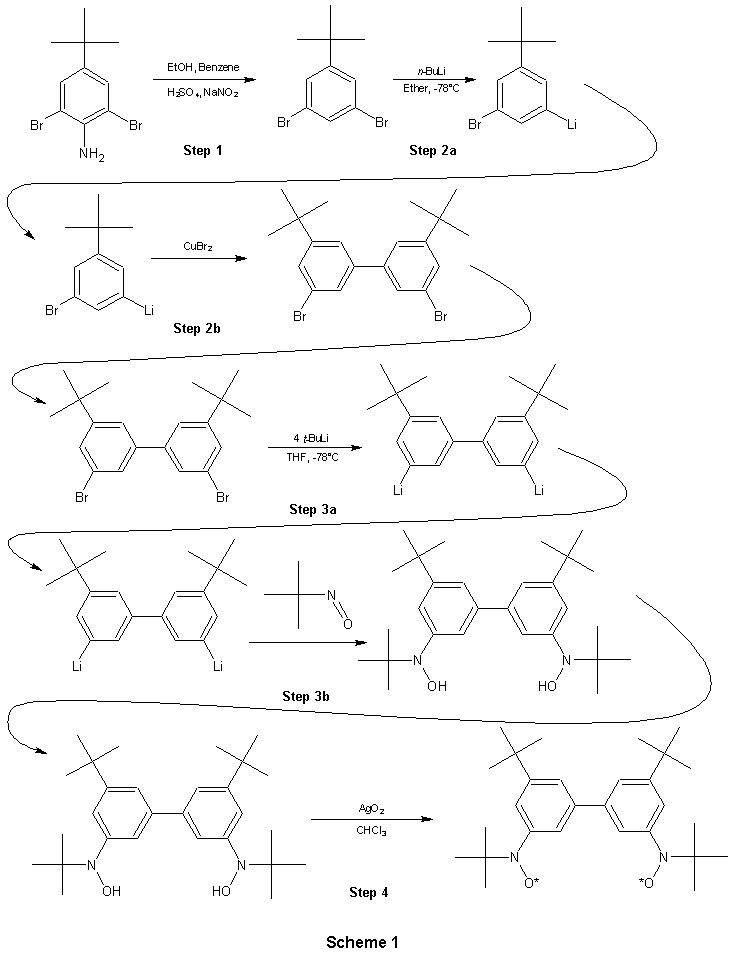
Although the synthesis of compound 3 was successful, the process
was not easy and took almost the whole ten weeks to complete. On
the following page is the synthesis scheme (Scheme 1) used to create
this diradical. Despite the fact that the final compound is stable,
many of the steps along the way are very moisture sensitive and therefore
were performed under vacuum in special evacuatable reaction vessels using
a vacuum line, septums, needles and dry solvents (no moisture). In
particular, it was step 3 that I had trouble with, performing it four times
and successful only twice. This reaction is performed with
a very moisture sensitive compound, the t-BuNO compound, and is
easily affected by contact with air and light. I am unsure of the
exact problems encountered in the failed trials, but it was probably a
result of these sensitivities. In the process of this synthesis I
learned and performed many valuable skills including performing reactions
under vacuum, handling of dry solvents, handling t-BuLi and n-BuLi,
weighing air sensitive compounds under nitrogen, and how to run a column.
In addition to these skills, I also learned how to operate a NMR machine
(200, 300, and 500 MHz) which I used on a daily basis to identify and check
the purity of the compounds I synthesized after each step. These
skills I am sure will be an asset to me in my career ahead.
 Using the 300 and 500 MHz NMR, I was able to establish that I had successfully
formed the correct diradical. On the following page are two sets
of NMR data; the first, Figure 2, is the compound created after
step 3, a hydroxyl amine, and the starting material for the formation of
the diradical; the second, Figure 3, is the final product, the diradical
itself. By looking at Figure 2 we notice certain peaks that
identify the compound as the hydroxyl amine I needed. The first peak
we notice is at approximately 1.2 PPM. In fact, this peak is actually
two singlet peaks right next to each other as is shown by the expansion
next to them. These singlets are peaks from the t-Butyl groups
on the compound. Unfortunately, they are so close together we are
unable to identify which peak is caused by which t-Butyl group.
The second peak to notice can be found at approximately 7.0 PPM.
Because this peak falls in a region of the spectrum where we know aromatic
protons show peaks, we know that the three types of protons on the phenyl
rings cause this peak. The compound before this, the product of step
2, showed three separate triplets in the aromatic region, one triplet for
each different type of proton, but in this compound the three protons have
blended to show one large singlet that we see around 7.0 PPM. The
third peak that has relevance to this hydroxyl amine is the last and smallest
and can only be seen by enlarging the region of the spectrum from 7 to
9 PPM. Once enlarged we notice a very broad, low peak caused by the
protons attached to the two oxygens in the molecule. The reason this
peak is so small is that it can hydrogen-bond with any moisture (water)
that is in the solution, thus binding it and hindering any strong signal
from being seen. Please note that other peaks not mentioned are caused
by impurities, TMS (0.00 PPM), and chloroform (@7.26
PPM).
Using the 300 and 500 MHz NMR, I was able to establish that I had successfully
formed the correct diradical. On the following page are two sets
of NMR data; the first, Figure 2, is the compound created after
step 3, a hydroxyl amine, and the starting material for the formation of
the diradical; the second, Figure 3, is the final product, the diradical
itself. By looking at Figure 2 we notice certain peaks that
identify the compound as the hydroxyl amine I needed. The first peak
we notice is at approximately 1.2 PPM. In fact, this peak is actually
two singlet peaks right next to each other as is shown by the expansion
next to them. These singlets are peaks from the t-Butyl groups
on the compound. Unfortunately, they are so close together we are
unable to identify which peak is caused by which t-Butyl group.
The second peak to notice can be found at approximately 7.0 PPM.
Because this peak falls in a region of the spectrum where we know aromatic
protons show peaks, we know that the three types of protons on the phenyl
rings cause this peak. The compound before this, the product of step
2, showed three separate triplets in the aromatic region, one triplet for
each different type of proton, but in this compound the three protons have
blended to show one large singlet that we see around 7.0 PPM. The
third peak that has relevance to this hydroxyl amine is the last and smallest
and can only be seen by enlarging the region of the spectrum from 7 to
9 PPM. Once enlarged we notice a very broad, low peak caused by the
protons attached to the two oxygens in the molecule. The reason this
peak is so small is that it can hydrogen-bond with any moisture (water)
that is in the solution, thus binding it and hindering any strong signal
from being seen. Please note that other peaks not mentioned are caused
by impurities, TMS (0.00 PPM), and chloroform (@7.26
PPM).
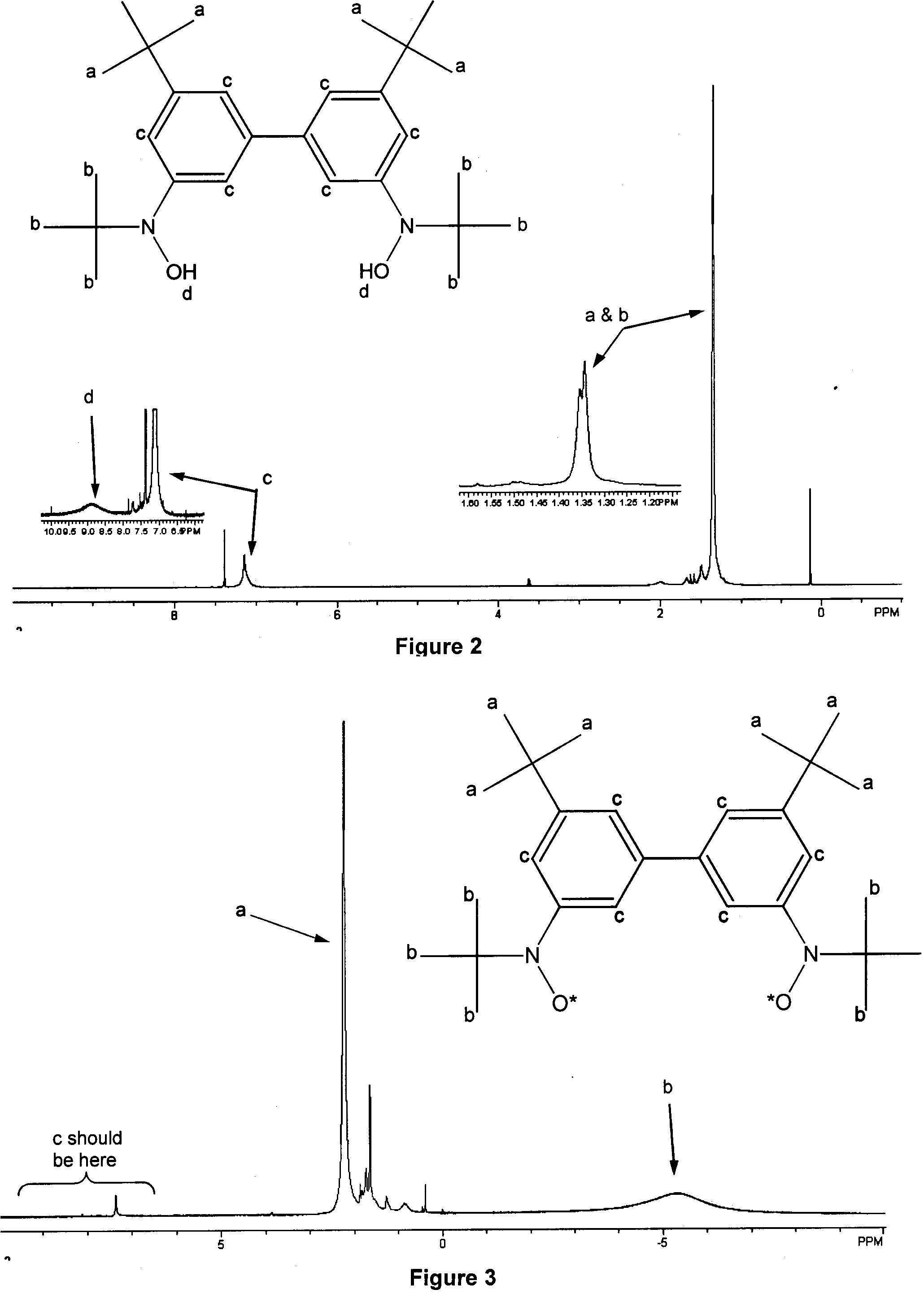
Once we have analyzed the NMR data from the hydroxyl amine, we can then
look at the NMR data taken for the product of step 4. There are several
indicators that this NMR was given by the diradical compound I was attempting
to synthesize. The most obvious indicator is that, other than the
chloroform peak, there is nothing in the aromatic region. This absence
of aromatic peaks is caused by the unpaired electrons interacting with
the aromatic protons, causing the aromatic protons to have a high spin
density and relax too fast for the NMR probe to detect them. The
interaction with the unpaired electrons also shifts them so far down field
(»-500 PPM) we are unable to see them
at all. The second thing to notice is the very low, broad peak
at approximately -5.3 PPM. This peak is caused by the protons on
the t-Butyl group attached to the nitrogen (and right next to the
radical sites). Again, the unpaired electrons are interacting with
these protons causing them to relax faster than normally, broadening the
peak and shifting it to below zero PPM. The third important peak
is the sharp singlet at approximately 2.2 PPM. This peak is
the signal from the other t-Butyl groups attached directly to the
phenyl rings. Because they are relatively far away from the radical
sites these t-Butyl groups are not affected as drastically by the
unpaired electrons. The effects that are seen are a result of the
unpaired electrons is a shift to the left, although it is a much smaller
shift compared to the t-Butyl groups attached to the nitrogen.
The reason for this difference in direction of shift and broadening are
explained briefly in the next paragraph.
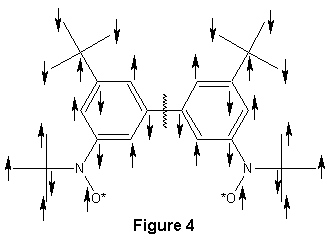
To explain the reason for the shifting of the t-Butyl peaks a little
background must be discussed beforehand. First, electron spins in
radicals (and all molecules) must be explained. In the external
magnetic field of the NMR instrument, every unpaired electron has a certain
spin direction. Because these unpaired electrons are in the p-orbitals
of the atoms they affect the electrons in p-orbitals on neighboring atoms
in such a way that the neighboring electrons spins are opposite that of
the unpaired electron. This opposite spin is then related to the
next electron in a similar way throughout the whole molecule as in Figure
4. (Note: A single arrow is placed between the oxygen and the
nitrogen because this is where the unpaired electron is, instead of on
just the oxygen or nitrogen.) The only exception in this molecule
is in between the two phenyl rings. Here we can see that the electrons
between the phenyl rings have the same spin, and therefore the unpaired
radical electrons also have the same spin, indicating a triplet state.
I have put my compound in this triplet state in order to explain the reason
behind the shifts in the NMR spectrum in Figure 3. Because
the NMR is taken while the sample is at room temperature there are larger
amounts of the compound in the triplet state than there are in the singlet
state. Only at very low temperatures will all the molecules in a
sample be in the singlet state. In the triplet state though, the
two phenyl rings act independent of each other allowing the two sides to
have the same direction spins on corresponding atoms. As a
result, the spins on the two different t-Butyl types are opposite,
and affect the shift of their peaks inversely i.e. one peak shifts right,
one peak shifts left.
In relation to the molecule being in the singlet and triplet state, it
would now be appropriate to discuss what testing will be done with my compound,
after I am gone. One of the first things that must be done is that
the cyclic molecule must be synthesized. A synthesis scheme has been
formed and seems fairly simple, but I am sure that it will also take several
weeks to create a usable sample. Once both compounds are attained
and are found to be pure, testing on energy levels will begin using ESR
(Electron Spin Resonance) spectroscopy and SQUID Magnetometry (Superconducting
QUantum Interference Device). Although I have very little idea of
how these machines work, or how to read the data from them, I do know that
when all results are received, the energy level difference between singlet
and triplet states will be found for both cyclic and acyclic molecules.
In order to do this, the molecules are cooled down to 2-5° K in order
to put all the molecules in a singlet state. The temperature is then
raised slowly until it is determined that a triplet state has been reached
and the energy difference between the two states can be determined.
After determining the energy difference between the singlet and triplet
states for both molecules, the theory of multiple pathways increasing the
energy difference by the same number of pathways will be proven or disproved.
If proven, this knowledge will greatly help chemists in the future in designing
organic magnets in order to take a place in our ever-growing magnetic world.
In conclusion, what I have done this summer was a miniscule part of this
on the edge research, but nevertheless, a vital part. Knowing the
energy differences between the different states is essential if organic
magnets are ever to take a place among our world. I am sure that
in time, we will not be using magnets made from iron, cobalt and manganese,
but instead will be integrating magnets made out of carbon, oxygen and
nitrogen in everything we do.
I must add, this summer has been a wonderful experience for me and I can
not imagine a better way of spending a summer. I have learned as
much chemistry in a summer as I did in a whole year of school. Not
only did I gain extra knowledge and skill in organic synthesis and testing,
but was also treated like one of the group members and given the opportunity
to present on a regular basis. I know that these skills will be an
asset as I continue in my education and on through my career. My
most sincere thanks to Dr. Andrzej Rajca and the whole Rajca Group.
References:
1. A. Rajca, Chemical Reviews, 1994, Vol. 94,
No. 4
2. A. Rajca, S. Rajca, J. Wongsriratanakul, Chem. Commun.,
2000, 1021-1022.
3. A. Rajca, S. Rajca, J. Am. Chem. Soc. 1996, 118,
8121-8126.
4. A. Rajca, K. Lu, S. Rajca, C.R. Ross II, Chem. Commun., 1999,
1249-1250.
5. D. A. Dougherty, Acc. Chem. Res., 1991, Vol. 24,
No. 3.
6. P. M. Lahti, 1995 ACS Symposium Series, Ó
1996, No. 644.

